



Exploring protein structure and protein sequences by making phylogenetic trees is not trivial but can be interesting and informative. It can be a productive way of deepening our understanding of the proteins and the relationships between them.
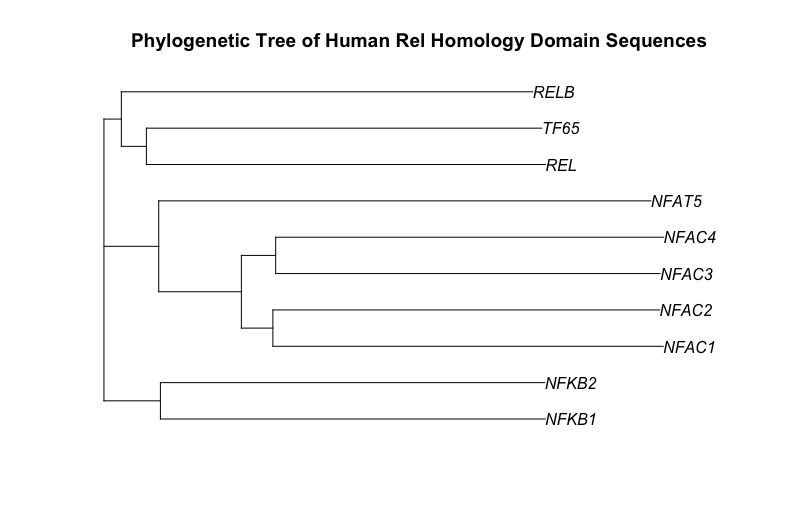
As a starting point for this blog post, I have chosen to draw a phylogenetic tree of the ten human proteins containing a rel homology DNA binding domain. This family includes the NF-kappaB family of transcription factors and the NFAT family of transcription factors. Both of these families change gene expression in the response to extracellular stimuli. The cytokine, tumor necrosis factor alpha (TNF), activates the NF-kappaB family of transcription factors. Stimulation of cells through antigen receptors, for example the T-cell receptor, activates the NFAT family of transcription factors.
Here is the phylogenetic tree that I have drawn:

It has been generated using the full length protein sequences which is an important point. The proteins are as follows (5 NF-kappaB family members & 5 NFAT family members):
- NF-kappaB subunit p105/p50 - P19838 (NFKB1_HUMAN)
- NF-kappaB subunit p100/p52 - Q00653 (NFKB2_HUMAN)
- NF-kappaB subunit Rel B - Q01201 (RELB_HUMAN)
- NF-kappaB subunit p65/Rel A - Q04206 (TF65_HUMAN)
- NF-kappaB subunit c-Rel - Q04864 (REL_HUMAN)
- NF-ATc1 - O95644 (NFAC1_HUMAN)
- NF-ATc2 - Q13469 (NFAC2_HUMAN)
- NF-ATc3 - Q12968 (NFAC3_HUMAN)
- NF-ATc4 - Q14934 (NFAC4_HUMAN)
- NFAT5 - O94916 (NFAT5_HUMAN)
The steps are as follows:
- Get the sequences into R. I’m using the package seqinr to extract protein sequences. The ways in which phylogenetics analysis is done is quite different depending on whether you are using DNA or protein sequences so it’s important to be aware of this.
- Perform a multiple sequence alignment. I’ve used the msa package. This process compares one sequence to another to another. There are different mathematical methods of doing this and different values that can be used within the different mathematical methods. The msa() function, by default, runs ClustalW with default parameters, one of the standard methods in biology. ClustalW has been around for many years. I first used it about 20 years ago when I did a short online course in bioinformatics. It can be used in my different tools including some web based interactive tools. The msa package also allows the use of the methods ClustalOmega or MUSCLE.
- Turn your alignment into a tree. This uses the alignment to calculate ‘distances’ between the proteins. The seqinr package has a method and the ape package is used too for neighbor-joining tree estimation. This is not a trivial matter as some changes between amino acids are more likely than others depending on the codon usage. Also some changes may lead a more dramatic change in function than others. Again, there are various algorithms.
- Draw your tree. This uses the ape package. For a small tree this is relatively simple but for larger trees there can be an artistic element to this. There are many options and a few are illustrated below.
Here is the script that I used to generate the tree above. There are other trees generated along the way...
SCRIPT START
### generating a phylogenetic tree of human Rel homology containing proteins
## packages required - may need downloading
# source("http://www.bioconductor.org/biocLite.R")
# biocLite("msa")
library(seqinr)
library(msa)
library(ape)
### Get the sequences into R.
# these are the accession numbers of the 10 proteins.
# for these 10 proteins I found them manually by searching the Uniprot database
accNo <- c("AC=P19838 OR AC=Q00653 OR AC=Q01201 OR AC=Q04206 OR AC=Q04864 OR AC=O95644 OR AC=Q13469 OR AC=Q12968 OR AC=Q14934 OR AC=O94916")
# using the seqinr package
# From Chapter 4 of the seqinr handbook.
# http://seqinr.r-forge.r-project.org/seqinr_3_1-5.pdf
### step 4.1 Choose a bank
choosebank() # show's available data bases
mybank <- choosebank(bank = "swissprot")
str(mybank)
# UniProt Knowledgebase Release 2016_08 of 07-Sep-2016 Last Updated: Oct 4, 2016
### step 4.2 Make the query
mybank <- choosebank(bank = "swissprot")
rel_seq <- query("relSeq", accNo)
# N.B. protein info NOT returned in the same order as requested
# returns list object with six parts
rel_seq$nelem # with 10 elements - thus 10 proteins
rel_seq$req # gives the five necessary names
### 4.3 Extract sequences of interest
rel_seqs <- getSequence(rel_seq)
# check the names of the sequences
getName(rel_seq)
# it is necessary to put the sequences in fasta format
# for the multiple sequence alignment.
# useful to store them locally so that's what this does
write.fasta(sequences = rel_seqs,
names = getName(rel_seq),
nbchar = 80, file.out = "relseqs")
### with sequences in a file we can open the file in the package msa
### read in Rel sequences from the file
mySeqs <- readAAStringSet("relseqs") # from package Biostrings
length(mySeqs)
# 10 sequences... which is correct
### Perform a multiple sequence alignment
myAln <- msa(mySeqs)
# this uses all the default settings and the CLUSTALW algorithm
myAln
print(myAln, show="complete")
### Turn your alignment into a tree
# convert the alignment for the seqinr package
myAln2 <- msaConvert(myAln, type="seqinr::alignment")
# this object is a list object with 4 elements
# generate a distance matrix using seqinr package
d <- dist.alignment(myAln2, "identity")
# From the manual for the seqinr package
# This function computes a matrix of pairwise distances from aligned sequences
# using similarity (Fitch matrix, for protein sequences only)
# have a look at the output
as.matrix(d)
# generate the tree with the ape package
# the nj() function allows neighbor-joining tree estimation
myTree <- nj(d)
# plot the tree
plot(myTree, main="Phylogenetic Tree of Human Rel Homology Domain Sequences")

# since all the species are human,
# it might be easier to read without the "_HUMAN"
# with the sub() function
myAln2$nam <- sub("_HUMAN", "", myAln2$nam)
# pile up the functions to make a new tree
tr <- nj(dist.alignment(myAln2, "identity"))
# plot the new tree
plot(tr, main="Phylogenetic Tree of Human Rel Homology Domain Sequences")
# from http://ape-package.ird.fr/ape_screenshots.html
plot(tr, "c")


plot(tr, "c", FALSE) # ignores edge length

plot(tr, "u", FALSE)

plot(tr, "f", FALSE, cex = 0.5)

"u", # unrooted - draws from the centre
font = 2, # makes font bold
edge.width = 2, # makes thicker lines
cex = 1.25, # increase font size a little
main = "Phylogenetic Tree of Human Rel Homology Domain Sequences")

# looks quite stylish but I'm not sure about ignoring edge length
plot(tr, "u",
use.edge.length = FALSE,
font = 2,
edge.width = 2,
main = "Phylogenetic Tree of Human Rel Homology Domain Sequences")

SCRIPT END
Resources and citations:
- seqinr package: http://seqinr.r-forge.r-project.org/
- msa package: https://bioconductor.org/packages/devel/bioc/vignettes/msa/inst/doc/msa.pdf
- msa package: U. Bodenhofer, E. Bonatesta, C. Horejsˇ-Kainrath, and S. Hochreiter (2015). msa: an R package for multiple sequence alignment. Bioinformatics 31(24):3997–3999. DOI: bioinformatics/btv494.
- ape package: http://ape-package.ird.fr/
- ape package: Paradis, E., Claude, J. and Strimmer, K. (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics, 20, 289–290
- ape package: Popescu, A.-A., Huber, K. T. and Paradis, E. (2012) ape 3.0: new tools for distance based phylogenetics and evolutionary analysis in R. Bioinformatics, 28, 1536-1537.
No comments:
Post a Comment
Comments and suggestions are welcome.